GO FAQs
These are our most frequently asked questions. If you don’t find your answer below, please contact us.
Browse the GO FAQ by topic:
- General: Overview
- General: Citing the GO
- Annotation
- Ontology
- PAN-GO Functionome
- AmiGO
- GO-CAMs
- Enrichment Analysis (EA)
- Subsets (slims)
- Mappings
- File Formats
- Database Access
General: Overview
What is the GO?
The Gene Ontology (GO) knowledgebase is a collaborative effort to address the need for consistent descriptions of gene products in different databases. The GO collaborators are developing three structured, controlled vocabularies (ontologies) that describe gene products in terms of their associated biological processes, cellular components and molecular functions in a species-independent manner. There are three separate aspects to this effort: first, we write and maintain the ontologies themselves; second, we make cross-links between the ontologies and the genes and gene products in the collaborating databases; and third, we develop tools that facilitate the creation, maintenance and use of ontologies.
The use of GO terms by several collaborating databases facilitates uniform queries across them. The controlled vocabularies are structured so that you can query them at different levels: for example, you can use GO to find all the gene products in the mouse genome that are involved in signal transduction, or you can zoom in on all the receptor tyrosine kinases. This structure also allows annotators to assign properties to gene products at different levels, depending on how much is known about a gene product.
What are all the possible uses of GO?
It would be impossible to list all the potential applications of GO, but applications for which GO has already been used include the following:
- integrating proteomic information from different organisms;
- assigning functions to protein domains;
- finding functional similarities in genes that are overexpressed or underexpressed in diseases and as we age;
- predicting the likelihood that a particular gene is involved in diseases that haven’t yet been mapped to specific genes;
- analysing groups of genes that are co-expressed during development;
- developing automated ways of deriving information about gene function from the literature;
- verifying models of genetic, metabolic and product interaction networks.
For references to these and other studies that have used GO, see the GO and the scientific literature page.
Why do we need GO?
To ask meaningful questions, biologists often need to retrieve and analyse data from disparate sources. For example, if you were searching for new targets for antibiotics, you might want to find all the gene products that are involved in bacterial protein synthesis, but that have significantly different sequences or structures from those in humans. But if one database describes these molecules as being involved in ‘translation’, whereas another uses the phrase ‘protein synthesis’, it will be difficult for you - and even harder for a computer - to find functionally equivalent terms.
The Gene Ontology (GO) knowledgebase is a collaborative effort to address the need for consistent descriptions of gene products in different databases. The GO collaborators are developing three ontologies - a word used by computer scientists to mean ‘specifications of a relational vocabulary’ - that describe biological processes, cellular components and molecular functions in a species-independent manner.
Ontologies provide a vocabulary for representing and communicating knowledge about a topic, and a set of relationships that hold among the terms of the vocabulary. They can be structurally very complex, or relatively simple. Most importantly, ontologies capture domain knowledge in a way that can easily be dealt with by a computer . Because the terms in an ontology and the relationships between the terms are carefully defined, the use of ontologies facilitates making standard annotations, improves computational queries, and can support the construction of inference statements from the information at hand.
Genomic sequencing projects and microarray experiments alike produce electronically-generated data flows that require computer accessible systems to work with the information. As systems that make domain knowledge available to both humans and computers, bio-ontologies such as GO and the many other bio-ontologies being created (see the OBO web page for some examples) for are essential to the process of extracting biological insight from enormous sets of data.
Which biological domains are supported by GO?
The current ontologies of the GO are molecular function, biological process, and cellular component. The ontologies are developed to include all terms falling into these domains without consideration of whether the biological attribute is restricted to certain taxonomic groups. Therefore, biological processes that occur only in plants (e.g. photosynthesis) or mammals (e.g. lactation) are included. Other biological ontologies are discussed in the OBO web site.
What is the PAN-GO Functionome?
The PAN-GO functionome is the set of all annotated functional characteristics (“Gene Ontology annotations”) for human protein-coding genes, as determined by the PAN-GO (Phylogenetic ANnotation using Gene Ontology) project, a collaboration between the Gene Ontology Consortium and the PANTHER evolutionary tree resource. Each annotation is the result of expert biologist review, based on integration of experimental data for related genes using explicit evolutionary modeling.
A detailed description of the process used to create the PAN-GO functionome, as well as an analysis of its contents, has been published in: Feuermann et al., A compendium of human gene functions derived from evolutionary modelling, Nature 2025.
If you are working on human protein-coding genes, we recommend you use the PAN-GO Functionome. It is designed to be as accurate and comprehensive as possible, while also being concise (a minimally redundant set of GO terms for each gene).
What is GO “content”?
GO content refers to the content of the ontologies themselves and the biology underlying it. It includes anything to do with terms and their organisation, definitions, synonyms and the relationships between terms.
I want to use GO, but I don’t know where to begin…
There are a number of possibilities for how researchers can make use of the GO.
The Gene Ontology website (http://geneontology.org/) is a very good place to begin learning about our knowledgebase, how GO resources are produced, and how we maintain them. It also illustrates how the research community most commonly makes use of these resources and how they can contribute. Exploring the items under the “Ontology” and “Annotations” on the menu will provide you with a very informative overview.
For more detail, please consult the open access The Gene Ontology Handbook, available online and as a downloadable PDF. As well as GO best practices and a discussion on the meaning of “function”, this text covers everything from introducing ontologies, to using GO resources in Python, and even how GO and similar ontologies may be used in clinical settings.
What am I allowed to do with the data?
The use and license of all GO data, software, and materials are covered on the Use and license page.
How is the GO used in genome analysis?
Functional annotation of newly sequenced genomes: Genome and full-length cDNA sequence projects often include computational (putative) assignments of molecular function based on sequence similarity to annotated genes or sequences. A common tactic now is to use a computational approach to establish some threshold sequence similarity to a SWISS-PROT sequence. Then the GO associations to the SWISS-PROT sequence can be retrieved and associated with the gene model. Under the GO guidelines, the evidence code for this event would be ‘inferred from electronic annotation’ (IEA).
Functional groupings of gene products: One aspect of the use of the GO for annotation of large data sets is the ability to group gene products to some high level term. For example, while gene products may be precisely annotated as having role in a particular function in carbohydrate metabolism (i.e., glucose catabolism), in the summary documentation of the data set, all gene products functioning in carbohydrate metabolism could be grouped together as being involved in the more general phenomena ‘carbohydrate metabolism’. Various sets of GO terms have been used to summarize experimental data sets in this way. The expectation is that published sets of high-level GO terms used in genome annotations and publications will be archived at the GO site. Some of these ‘GO slims’ are already available.
General: People & Funding
How can I contribute to GO?
We welcome your contributions!
The GO knowledgebase is constantly evolving, and we welcome feedback from all users. Research groups may contribute to the GOC by either providing suggestions for updating the ontology (e.g. requests for new terms) or by providing annotations, that is, associations between genes or gene products and ontology terms. Suggested edits are reviewed by the ontology editors and implemented where appropriate.
- To suggest updates or modifications to the ontology, please visit our documentation on contributing to the ontology.
- To learn more about the best approach to contributing GO annotations, please visit our documentation on contributing annotations.
Please be sure to contact the GOC before carrying out any annotation work you intend to submit; this will ensure that GOC mentors and trainers can be of assistance in producing data sets in agreement with the GOC annotation policies and format requirements.
Who makes up the GO Consortium?
Back in 1998, the Gene Ontology began as a collaboration between three model organism databases, namely Flybase (Drosophila), the Saccharomyces Genome Database (SGD), and the Mouse Genome Database (MGD). Today, the GO Consortium is formed by many databases, including several of the world’s major repositories for plant, animal, and microbial genomes. Visit this page to see a complete list of member organizations of the Gene Ontology Consortium.
What is the difference between GO, GOC, GO Central, and GOA?
GO, GOC, GO Central, and GOA are similar but not quite interchangable. Here’s a quick breakdown of these commonly confused acronyms:
GO
- Gene Ontology: Although formally we use GO to refer to the entire GO knowledgebase, this can also refer to just the ontology. In addition to releases of data products like the ontology, annotations, and GO-Causal Activity Models (GO-CAMs), the GO provides the AmiGO browser and many other tools.
GO Central
- The GO Central project is embedded in the larger GO Consortium and circle of GO Collaborations. Within this complex landscape of resources, GO Central has several activities, including ontology development, tool development, annotation support, quality assurance, and outreach.
GOC
- Gene Ontology Consortium: The databases, labs, and other groups that officially participate in the maintenance of the GO knowledgebase. See a complete list of member organizations of the Gene Ontology Consortium.
GOA
- EBI’s Gene Ontology Annotation (GOA) Database, one of many of GO’s contributing groups, is the EMBL-EBI project responsible for many of the GO annotations found in UniProt, RNACentral, and Complex Portal as well as the QuickGO browser.
How do I become a member of the GO Consortium?
The most important criterion for GO Consortium membership is that the members contribute something to the collection of resources that we make available to the public (almost all members contribute annotations; several contribute to the ontologies; a few contribute software). The scientists involved in working with GO in these member groups communicate via the GO mailing lists and GitHub to discuss development issues in the ontologies. If you represent a database that wishes to join the GO Consortium please contact the GOC.
Anyone with a more general interest in the GO should follow our Bluesky (@geneontology.bsky.social) to receive updates about the GO.
Who funds GO?
Direct support for the Gene Ontology Consortium is provided by an R01 grant from the National Human Genome Research Institute (NHGRI) [grant HG02273]. Funding for the GO member organizations is detailed on our annotation contributors page.
General: Citing the GO
How do I cite the GO?
Citation information for the Gene Ontology can be found on the GO Citation Policy page.
What is the minimum information to include in a functional analysis paper?
Most journals require authors to submit high-throughput data to public repository as a prerequisite for publication. As part of this process, the methods used to analyse data need to be reported in detail; this applies to both statistical and functional analysis. For papers describing enrichment analysis using GO, this means that the methods section should include the following information, to ensure the analysis is reproducible (an important criteria for reviewers’ approval):
- What analysis tool was used and what version
- What statistical analysis method was applied, and what correction factors were applied if any
- Date/release version of both the GO ontology file and the GO annotation file used
- Background genome/proteome/dataset used in the analysis, including strain if applicable
- Whether any enriched terms were excluded from the results due to low numbers of query genes associated with the term (e.g., if you only included GO terms in the results which have more than 3 query genes)
- Please cite the appropriate GO papers
The supplemental data files should include:
- List of the IDs used and the IDs which were rejected by the analysis tool, if any
- Full list of enriched terms
When undertaking the functional analysis and interpreting the results, consider:
- Is the number of genes analysed statistically valid? Or is the number too small to observe enrichment, or too large for the enrichment to be meaningful. (E.g., for a microarray experiment with ~25,000 interrogated transcripts, it would be difficult to observe enrichment when analysing less than 150 IDs; if the list were longer than 3,000 IDs, clearer results would require further filtering, e.g. based on significance threshold or fold change. This is only a rough guide.)
- Consider using more than one functional analysis tool, as well as fine-tuning the parameters used. You may also wish to look at overlap between results from different approaches.
- Think about the biology. E.g., if you need/wish to make a choice among enriched terms to show in a summary table, use descriptive GO terms. Do not only pick terms you’re particularly interested in, and consider that very broad or generic terms, such as ‘metabolic process’, can be uninformative.
Annotation
What is annotation?
What does it mean to do GO annotation of genes or proteins? Terms from the Gene Ontology are applied in the annotation of gene products or protein complexes in biological databases. GO annotations are associations made between gene products or protein complexes and the GO terms that describe them. An annotation also includes an evidence code and a reference that supports the gene product/term association.
How do I submit annotations to GO?
We welcome your contributions!
We welcome contributions to the Gene Ontology knowledgebase, both in terms of annotations and for feedback and additions to the ontology.
Before making contributions, we recommend that you contact the Gene Ontology Consortium (GOC) before annotation work is carried out; this will ensure that GOC mentors and trainers can be of assistance in producing data sets in agreement with the GOC annotation policies and format requirements.
To learn more details, visit the page on contributing to GO.
What is a ‘gene product’?
GO uses the term ‘gene product’ to refer collectively to genes and any entities encoded by the gene, e.g. proteins and functional RNAs.
How are gene products associated with GO terms?
A gene product can be annotated to zero or more nodes of each ontology, at any level within each ontology; annotation of a gene product to one ontology is independent of its annotation to other ontologies. Annotations should reflect the normal function, process, or localization (component) of the gene product; an activity or location observed only in a mutant or disease state is therefore not usually included. The member databases of the GO Consortium use manual and automated methods to annotate genes or gene products using GO terms. Both manual and automated annotations are made according to two principles: first, every annotation must be attributed to a source, which may be a literature reference, another database or a computational analysis; second, the annotation must indicate what kind of evidence is found in the cited source to support the association between the gene product and the GO term. GO uses a simple controlled vocabulary to indicate the type of evidence found in the cited reference to support the annotation.
Can a single gene product be annotated with more than one GO term?
Yes!
It is possible and usually expected for a single gene/gene product to be associated with more than one GO term. The fact that you may have found that there are two or more different GO terms associated with a single gene/gene product in your results should not be a cause for concern.
The Gene Ontology allows users to describe a gene/gene product in detail, considering three main aspects: its molecular function, the biological process in which it participates, and its cellular location.
For example, the homeobox D9a gene product from zebrafish has numerous GO terms associated with it. Each term describes details about this gene’s molecular function, localization in the cell, or its involvement in certain biological processes. One GO term explains that this gene product carries out the molecular function of selectively interacting with DNA (DNA binding), while a different GO term explains that this gene product is found in the nucleus of the cell.
Trying to write one single term that describes in detail everything about a gene/gene product in a single statement would require the existence of as many terms as genes there are - for all species - in the planet. This would be very unpractical and not easily scalable. Instead, the use of ontologies help us organize information in a way that allows researchers to use the same term to describe a characteristic that is shared by more than one gene product (e.g. all the genes involved in the process ‘translation’), and more than one term to describe all the characteristics of each gene product, as in the example above. This is a reason why you would see more than one GO term associated to a single gene/gene product.
Can a gene or gene product be annotated to more than one term from an ontology?
Yes, a gene product can be annotated to zero or more nodes of each ontology, at any level within each ontology. See the introduction to GO annotations for more information.
Why are some gene products annotated to both a parent term and a child term?
This is done when there is explicit evidence to support separate annotations; usually it means that there is strong evidence for a more general annotation (parent term) and weaker evidence supporting a more specific annotation (child term).
From the GO annotation guide:
Uncertain knowledge of where a gene product operates should be denoted by annotating it to two nodes, one of which can be a parent of the other. For instance, a yeast gene product known to be in the nucleolus, but also experimentally observed in the nucleus generally, can be annotated to both nucleolus and nucleus in the cell component ontology. Even though annotation to nucleolus alone implies that a gene product is also in the nucleus, annotate to both so as to explicitly indicate that it has been reported in the two locations. The two annotations may have the same or different supporting evidence.
What is an evidence code?
Every annotation must be attributed to a source, which may be a literature reference, another database or a computational analysis. The annotation must indicate what kind of evidence is found in the cited source to support the association between the gene product and the GO term. A simple controlled vocabulary is used to record evidence; and the evidence codes are simply the three-letter codes used to signify the type of evidence cited. More information on the meaning and use of the evidence codes can be found in the GO evidence codes documentation.
Where can I view or download the complete sets of GO annotations?
Annotations from GO Consortium member groups can be downloaded here. These are taxon-specific, although some have multiple taxons in the file. For example, the CGD GAF includes several Candida as well as Debaryomyces hansenii and Lodderomyces elongisporus.
If your organism is included in a multi-organism file, and you would like to extract just your organism of interest, you can filter by taxon. Verify your taxon ID and refer to the file format guides under “Types of GO annotation files” to determine which column to sort on.
If your organism is not available from our downloads page, you can use AmiGO to view or download annotations. Filter for your taxon under “Organism”; AmiGO allows users to download up to 100,000 annotations. QuickGO is EMBL-EBI’s GO browser and has a download limit of 2,000,000. Please see our FAQ on AmiGO and QuickGO data, software to browse the GO and finding species that are not available in AmiGO.
How do I access older versions of gene association files?
For files produced after 2018-03-06, please use the /annotation directory from http://release.geneontology.org/. GO annotations from 2018-03-06 to present are also accessible through the DOI-versioned release stored in Zenodo.
For files older than 2018-03-06, there is some data that is no longer maintained on our archive: http://archive.geneontology.org/full/ Please note, even though some files appear to be recently updated, this archive is no longer maintained and may refer to URLs that are no longer active.
If you do not find the release you are looking for, please contact us.
How do I find all annotations for species X that I can’t find in AmiGO?
Open QuickGO. Click on the “View GO annotations” box located beneath the search box. This will lead you to an Annotation browser page where you can filter icon based on taxon, evidence, GO term, gene product and more.
Click on taxon to input the taxon identifier of the species you would like to get GO annotations for (Example: 6279 for Brugia malayi). You can input more than one taxon.
Click Apply to get your results. Use the Statistics link to see the breakdown of the different annotations including Annotations per Reference, Gene products per Evidence, Annotations per Aspect, Gene products per GO ID, etc.
Sometimes the number of GO annotations changes significantly over a short period of time. Why?
Most annotations in association files are electronically inferred (IEA). As with all types of annotations, IEAs change over time, with an overall increasing trend. However, in the specific case of IEAs, significant fluctuations in numbers may sometimes be observed over a short period of time. Nearly always, these are not due to bugs, but rather to the following reasons and/or to a combination thereof:
- All IEA annotations that are over one year old are removed from association files. This is part of quality control procedures. Another procedure the GO started implementing in mid-2014 are taxonomic checks.
- Electronic annotations are provided to UniProt-GOA by various groups, including Ensembl, InterPro and UniProt. UniProt-GOA then includes these in their annotation files that they submit to the GO Consortium. There are numerous reasons why electronic annotations can fluctuate; e.g., InterPro may have changed a mapping that affected a large number of annotations; a mapping between a GO term and a UniProt keyword may have been added or removed; Ensembl may have changed their orthology sets; new quality checking procedures may have been introduced; a supplying group may have had a problem providing the annotations. Since electronic annotations tend to hit a large number of proteins, it is more likely to observe larger fluctuations than one would in a manual annotation set. UniProt-GOA aims to record all the known changes to the datasets they provide in the release notes here: http://www.ebi.ac.uk/GOA/news
- New genome assemblies for various species are periodically released, and that may contribute to changes in gene annotations.
- Changes are good. Our knowledge foundation is growing and increasing and information is continuing to be added based on existing, older literature.
- Relevant paper: Understanding how and why the Gene Ontology and its annotations evolve: the GO within UniProt.
However, if you think that an observed change in the size of an annotation file cannot be explained by any of the above, and suspect a bug, please contact us using our Contact Form.
Why have the number of electronic annotations (IEA) changed in 2026?
The GO Consortium is updating how we generate electronic annotations to improve accuracy and coverage. As part of aligning the EBI-GOA (QuickGO) and GO Central (AmiGO) pipelines, we’ve made several changes that affect the electronic annotations (IEA):
Key changes in 2026:
- Keywords2GO pipeline retired for most species, but remains active for viruses (NCBITaxon:10239)
- New annotation sources added from PAN-GO, TreeGrafter, and ARBA projects
What to expect: You may notice increases or decreases in IEA counts for specific organisms as we optimize annotation quality. These changes reflect our commitment to providing the most accurate and valuable GO annotations.
If you notice significant changes that affect your work, please contact us with specific details about the organism and annotation changes you’ve observed.
Note: Additional pipeline updates will continue throughout 2026.
What is the difference between the filtered_goa_uniprot_all.gaf and filtered_goa_uniprot_all_noiea.gaf?
Why can’t I find my organism in the GOA UniProt_All gene association files, doesn’t this GAF contain all annotations?
First of all, note that GOA≠GO; the Gene Ontology Annotation (GOA) Database is a contributor to the Gene Ontology knowledgebase and active member in the Gene Ontology Consortium, but is a different entity from the GO.- see our FAQ on GO, GOA, GOC, and GO Central. The files in question, using the current archive as an example, are:
- http://current.geneontology.org/annotations/filtered_goa_uniprot_all.gaf.gz
- http://current.geneontology.org/annotations/filtered_goa_uniprot_all_noiea.gaf.gz
The upstream file for both of these is https://ftp.ebi.ac.uk/pub/databases/GO/goa/UNIPROT/goa_uniprot_all.gaf.gz. We’ll refer to this as the “upstream” file. Also, archived releases up to and including 2023-04-01 refer to these files by the legacy names goa_uniprot_all.gaf.gz, goa_uniprot_all_noiea.gaf.gz, goa_uniprot_all_noiea.gpad.gz, goa_uniprot_all_noiea.gpi.gz. We renamed these files in April 2023 to clarify the contents.
filtered_goa_uniprot_all.gaf
For filtered_goa_uniprot_all.gaf, the GO takes the upstream file and removes all of the “canonical” species from it, essentially filtering out all species that have annotations that are provided by another resource included in the GO annotation dataset. For example, we ingest and process SGD’s data (https://sgd-prod-upload.s3.amazonaws.com/latest/gene_association.sgd.gaf.gz) for NCBITaxon:285006, NCBITaxon:307796, NCBITaxon:41870, NCBITaxon:4932, and NCBITaxon:559292, so we filter out these five taxa from the upstream file for our own version of this file. This is repeated for every resource we ingest, leaving us with a file that is the remainder of what is not otherwise spoken for in the GO annotation set.
goa_uniprot_all_noiea.gaf
This is the same as the filtered_goa_uniprot_all.gaf.gz file, except with electronically inferred (IEA) annotations filtered out. This is the file that is included in the AmiGO load and available at http://amigo.geneontology.org.
What criteria are used to annotate genes with GO terms?
A variety of criteria are used for each annotation including experimental results, sequence similarity and curator judgement. See the GO annotation guide for more information.
How is annotation quality controlled to ensure consistency between databases?
The accuracy of GO annotations is a high priority for all members of the GO Consortium. Each member organization is responsible for keeping its own annotations accurate and up to date, and for correcting any errors. Users can report errors to the GO helpdesk; any comments on annotations will be forwarded to the appropriate contributing group.
The GO Consortium is also looking into possible ways to improve quality assurance further, such as manually reviewing selected annotations and developing tools to automate detection of potentially erroneous annotations.
What are the advantages and disadvantages of manual annotation?
The most reliable annotations are those made manually by database curators based on primary and occasionally on review literature. Manual annotations usually cite experimental evidence that provides strong support for the association of a GO term with a gene product, and can be done at a very detailed level. The chief disadvantage of manual annotation is that it is labor-intensive, requiring a lot of time and effort from trained biologists.
What are the advantages and disadvantages of automatic annotation?
One advantage of automatic annotation is speed: wholly or partially automated methods facilitate the annotation of much larger sets of known or predicted gene products than can be produced manually. Automated annotation methods generally yields more broad (less detailed) annotations compared to manual annotation.
How often does automatic annotation give results that are consistent with manual annotation?
In general, electronic annotations are rarely incorrect, as they are annotations to very high-level GO terms. For example, the GOA group at EBI reports:
Usually manual annotation simply provides deeper-level terms in GO. In 93% of cases GOA’s electronic annotation is in the same GO lineage as the manual annotation. Some users have used our manual annotation to assess the quality of their automatic GO annotation techniques. They have found a few manual annotation errors by Proteome Inc. but no errors (so far) of manual annotation by Swiss-Prot staff have been reported to GOA. A few InterPro2GO errors have been reported but not very many. So, in general, our electronic techniques are very accurate, and are sometimes based on manual annotation. For example, Swiss-Prot keywords are usually manually annotated to Swiss-Prot entries; by using a mapping of Swiss-Prot keywords to GO, GOA inherits the high quality of Swiss-Prot manual annotation.
Text from Clark, et al., 2005
“The quality of electronic annotation has recently been assessed in some detail (Camon et al., 2005). This research found that in the worst case scenario, the generation of electronic annotations using the interpro2go, spkw2go, and ec2go mapping files precisely predicted the correct GO term 60% to 70% of the time, with the remainder of the predictions being to insufficiently specific GO terms. The high precision was found to be due to the basing of electronic annotations on manually curated mapping files. Curators noted that it was more important for database curation to be accurate than to have complete coverage, and the figures above demonstrate that this is the tendency with electronic annotation.”
How are binary interactions curated by the IntAct group selected for export to GO as protein binding (GO:0005515) annotations?
All binary interactions evidences in the IntAct database, including those generated by Spoke expansion of co-complex data, are clustered to produce a non-redundant set of protein pairs. Each binary pair is then scored, using a simple addition of the cumulated value of a weighted score for the interaction detection method and the interaction type for each interaction evidence associated with that binary pair. Only experimental data is scored, inferred interactions, for example, would not be scored, and any low confidence data, or data manually tagged by a curator for exclusion from the process, are also not scored. Isoforms and post-processed protein chains are regarded as individual proteins for scoring purposes. Score weightings were determined using the PSI-MI CV.
Once the interactions have been scored, a cut-off filter of 9 has been established, below which the interaction is not exported to UniProtKB and to the Gene Ontology annotation files. Additional rules ensure that any protein pair scoring above 9 must also include at least one evidence of a binary pair, excluding spoke expanded data, before export to UniProtKB/GOA.
These criteria ensure that:
* Only experimental data is used for making the decision to export the protein pair to UniProtKB/GOA as a true binary interacting pair
* The export decision is always based on at least two pieces of experimental data. A single evidence cannot score highly enough to trigger an export
* An export cannot be triggered if the protein pair only ever co-occurs in larger complexes, there must be at least one evidence that the proteins are probably in physical contact.
IMEx will only call an interaction ‘direct’ when performed with 2 purified molecules in vitro so any method using whole cells or cell lysates would not be regarded as direct. The described methodology will give you binary i.e. either direct or involved in the same small complex.
For more details, please see the corresponding entry on the the IntAct FAQ page.
Where can I find software to allow me to make or edit GO annotations?
GO’s Noctua tool is a curation platform that can be used to make GO annotations as well as GO Causal Activity Models (GO-CAMs). See an overview on the Tools page.
GO annotations can also be made and edited using various database-specific tools. Please contact the relevant database to find out how their GO annotation is done. The GMOD online tool, Canto, supports functional gene annotation by community researchers as well as by professional curators.
How do I annotate a novel genome with GO annotations?
Currently, GO recommends groups submit their transcriptomes to NCBI. These submissions will reach UniProt, where InterPro2GO automatically creates GO annotations. These annotations, made with the IEA evidence codes (Inferred from Electronic Annotation), will be made available in a future GO release.
GO does not recommend groups create their own IEAs with internal tools due to reproducibility and accuracy concerns.
What gene or protein IDs should I use?
The list of authoritative database groups for certain species lists the database groups who assume sole responsibility for collecting and submitting annotations for one or more species. If you can convert your IDs into the IDs used by that database group, you will be able to find the data you are looking for far more quickly and efficiently.
We maintain a list of suggested resources for mapping gene and protein IDs.
I have a question about gene or protein nomenclature
The GO Consortium is not involved in naming genes at all, in any organism. The GO vocabularies describe attributes of gene products; they are not collections of gene names or protein names.
Gene names are generally standardized within an organism but not necessarily between organisms (with some notable exceptions, such as the ongoing effort to make human and mouse gene names consistent). We suggest that you direct your query to the database or nomenclature committee for your organism. For example, human gene names are maintained by the HUGO Gene Nomenclature Committee (HGNC), mouse gene names by MGI, etc.
Ontology
What is an ontology?
Ontologies are ‘specifications of a relational vocabulary’. In other words they are sets of defined terms like the sort that you would find in a dictionary, but the terms are given hierarchical relationships to one another. The terms in a given vocabulary are likely to be restricted to those used in a particular field or domain, and in the case of GO, the terms are all biological.
How can I suggest new GO terms?
The GO vocabularies are updated on a regular basis, and suggestions from the community for additional terms or for other improvements are very welcome. Please visit the page on contributing to GO for details on how to submit your contributions.
Does the GO ID have any meaning?
GO IDs are unique identifiers, however, they do not encode any information about a term or its position relative to other terms in the tree. See more about GO terms.
Where can I find the number of terms in each of the ontologies?
You can find the number of terms on each of the ontologies two ways:
- Go to our Release statistics
- The Ontology section will have counts for Biological Process, Molecular Function, and Cellular Component terms.
- Go to AmiGO:
- Under the ‘Advanced Search’ section in the middle of the page, use the drop-down menu to choose “Ontology”. You don’t need to type anything on the ‘Quick search’ box.
- This action will send you to the ‘Information about Ontology search’ page. There, open the ‘Ontology source’ filter menu on the left.
As of January 2023 (2023-01-01), the number of terms on per ontology were:
- 27,942 Biological process
- 11,263 Molecular Function
- 4,043 Celular component
Please note that the number of terms changes between releases: new terms are created, existing terms may be merged, and some terms are obsoleted to reflect the current understanding of biology. This is why it is essential to provide a release version when citing the GO.
If you need a reference for this information, refer to our citation policy and license.
How can I calculate the “level” of a GO term?
GO terms do not occupy strict fixed levels in the hierarchy. Because GO is structured as a graph, terms would appear at different ‘levels’ if different paths were followed through the graph. This is especially true if one mixes the different relations used to connect terms.
A more informative metric would be the information content of the node based on annotations. See, for example, the work of Alterovitz et al..
Can a term in one ontology have parents in one of the other two ontologies?
Yes - there are links between molecular function, biological process, and cellular component ontologies. Note that any one term will ONLY have is_a parentage up to one of the root terms.
Can a term that is listed in two places in an ontology file have children in one place but not in the other?
No - the term will always have the same children wherever, and however many times it appears.
How are membrane proteins described in GO?
GO cellular component terms describe where a gene product is active, rather than describing the physical features of proteins. For membrane proteins, the topology of the protein or must be taken into account.
How GO represents membrane locations: Transmembrane and anchor proteins are annotated to the membrane itself, such as:
- plasma membrane (GO:0005886)
- nuclear membrane (GO:0031965)
- mitochondrial membrane (GO:0031966)
- endoplasmic reticulum membrane (GO:0005789)
Membrane sides: Proteins and protein complexes that are loosely bound to a membrane surface, but are not integrated into the hydrophobic region should be annotated to child terms of side of membrane (GO:0098552). These annotations are unique as they rely on topological (HMMs, known transmembrane domains or anchors, etc.) as well as experimental evidence. For example, a protein associated with the plasma membrane should be annotated to one of:
- cytoplasmic side of plasma membrane (GO:0009898)
- external side of plasma membrane (GO:0009897)
What you’ll see in GO annotations: Proteins are typically annotated to the most specific location term that the evidence supports. For example, if research shows a protein functions on the cytoplasmic side of the plasma membrane and the topology shows the protein is not transmembrane, it should be annotated to that specific term.
What GO doesn’t include: You won’t find terms like “type I membrane protein” in GO, because these describe how a protein is structured rather than where it’s located. Some older terms like “integral to membrane” have been updated to better reflect current understanding of cellular organization.
For researchers: When searching GO or interpreting results, keep in mind that GO annotations are based on available published evidence rather than providing comprehensive coverage of all protein functions and locations.
How do I get the term names for my list of GO IDs?
How do I get GO IDs for my GO terms?
What about GO definitions?
You can use the AllianceMine’s Upload List tool available at the Alliance website! This tool will return a table of GO ID, GO term name, and GO term description for each valid GO ID you supply. This will work for any organism, as the GO is the same!
- Go to the Upload List tool on AllianceMine
- In the List Type pull down, select
GO Term - Enter your GO ids or upload a file, making sure GO IDs have the correct format (GO:0016020, GO:0016301…)
- Click on
Continue, and then on the next page use theSave Listbutton. - You can use the
Save listbutton on the next page to use this list in AllianceMine, or use theExportbutton to see download options.
If you need the aspect (cellular component, molecular function, or biological process) for each term, you can add this to the results before saving. Use the Add Columns, click Namespace to highlight that option, then click the Add 1 columns button in the lower right. You can also use the AllianceMine features to filter your list, for example to select only molecular_function terms in your list.
If you have a list of GO terms and wish to retrieve GO IDs and/or definitions, you can use the steps above. Make sure multi-word GO terms are in double quotes (sporulation,”lactase activity”,”codeine metabolic process”) as the tool will otherwise recognise spaces as delimiters.
Can I download the ontologies as an Excel spreadsheet?
It is not possible to download the ontologies as a tabulated spreadsheet. The complex graph structure of GO, where terms can have one or more parent terms, makes it impractical to be rendered as a spreadsheet. It would probably also be too big for software packages such as Excel to cope with.
Where can I find software to allow me to edit the GO terms?
- Protege: Protege is a free, open-source ontology editor and framework for building intelligent systems. At this time, all ontology editors for GO use this program.
Where have the ‘unknown’ terms gone?
Good principles of ontological design state that terms should represent biological entities that actually exist, e.g., functional activities that are catalyzed by enzymes, biological processes that are carried out in cells, specific locations or complexes in cells, etc. To adhere to these principles the Gene Ontology Consortium has removed the terms ‘GO:0000004 biological process unknown’, ‘GO:0005554 molecular function unknown’ and ‘GO:0008372 cellular component unknown’ from the ontology. The “unknown” terms violated this principle of sound ontological design because they did not represent actual biological entities but instead represented annotation status. Annotations to “unknown” terms distinguished between genes that were curated when no information was available and genes that were not yet curated (i.e., not annotated).
Annotation status is now indicated by annotating to the root nodes, i.e. ‘GO:0008150 biological_process’, ‘GO:0003674 molecular_function’, or ‘GO:0005575 cellular_component’. These annotations continue to signify that a given gene product is expected to have a molecular function, biological process, or cellular component, but that no information was available as of the date of annotation. Adhering to principles of correct ontology design should allow GO users to take advantage of existing tools and reasoning methods developed by the ontological community.
PAN-GO Functionome
See also: What is the PAN-GO Functionome?
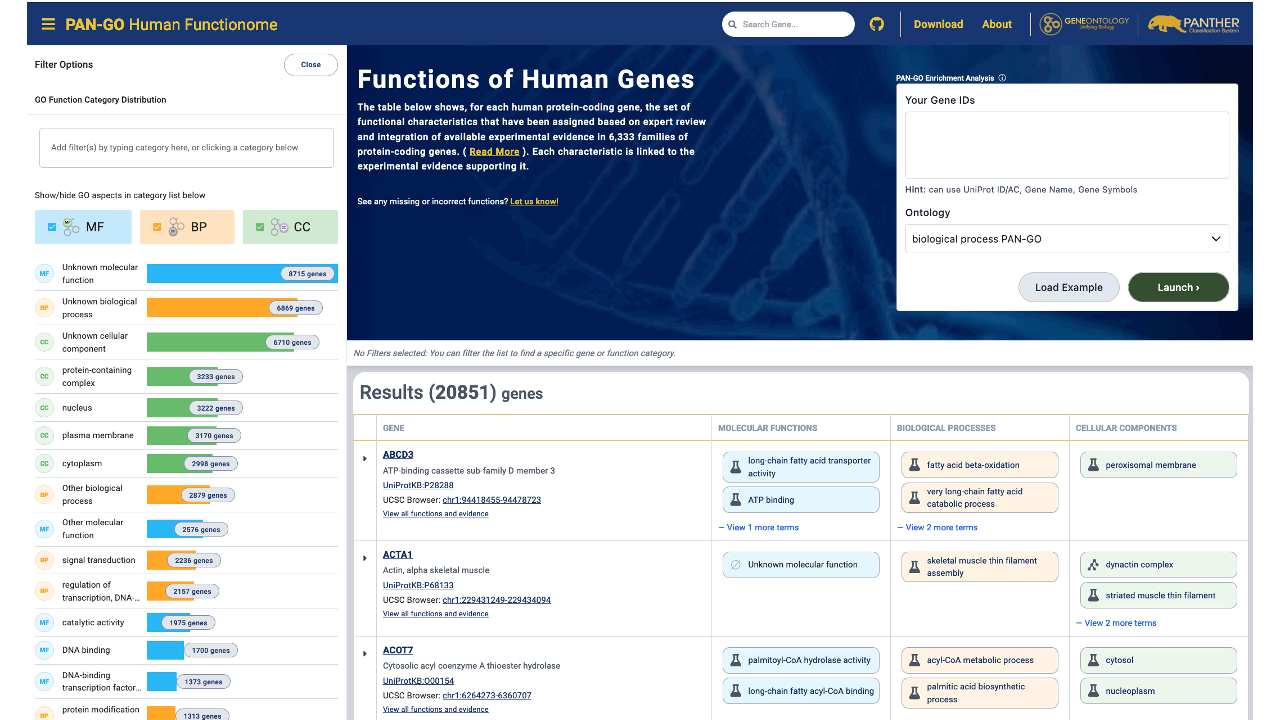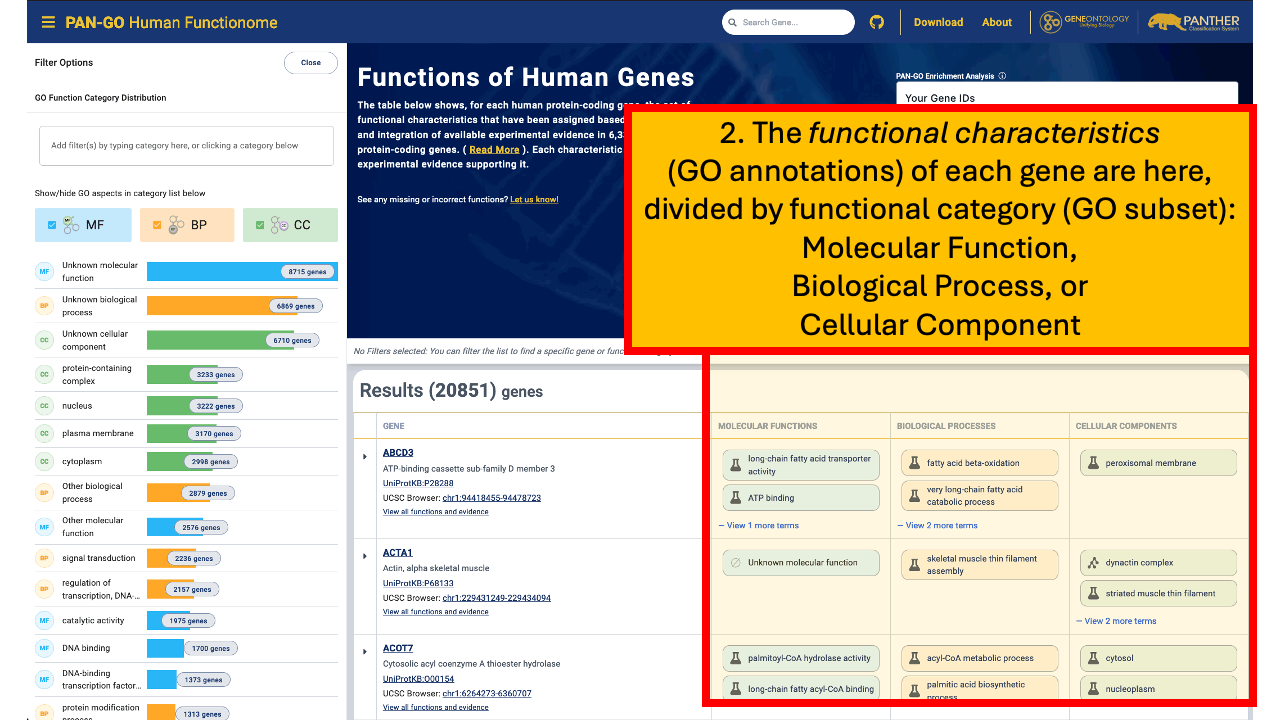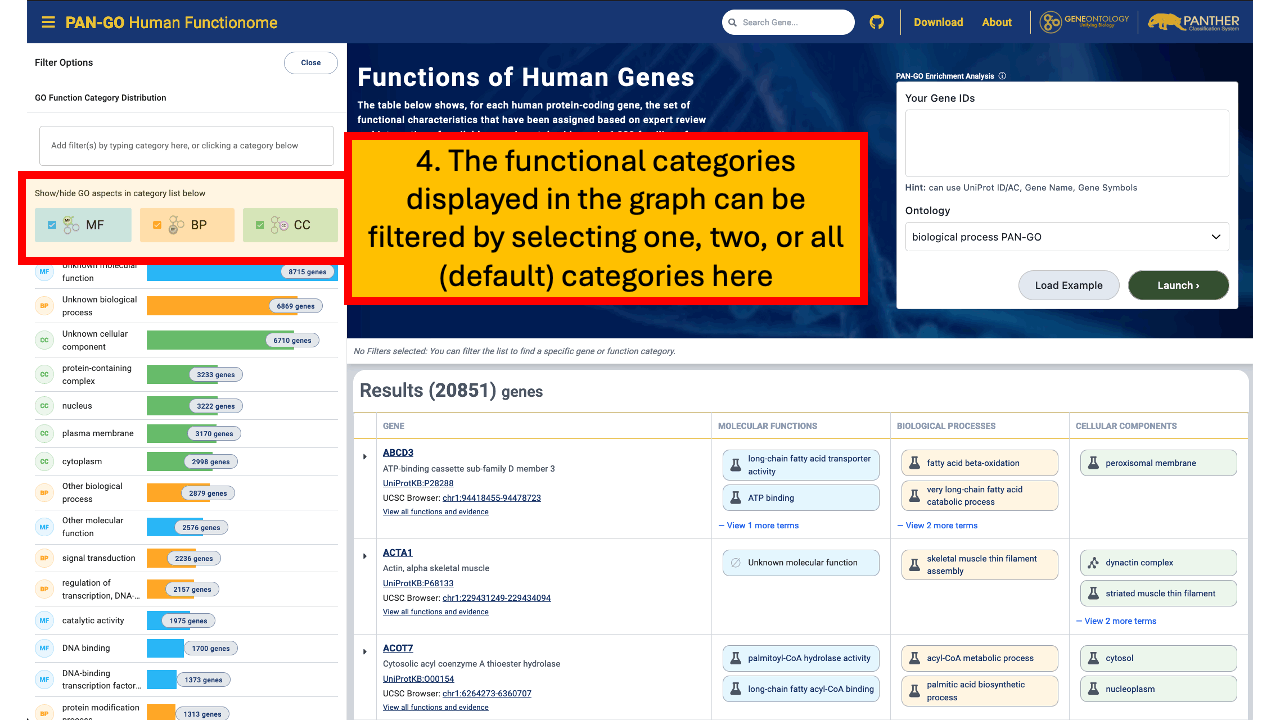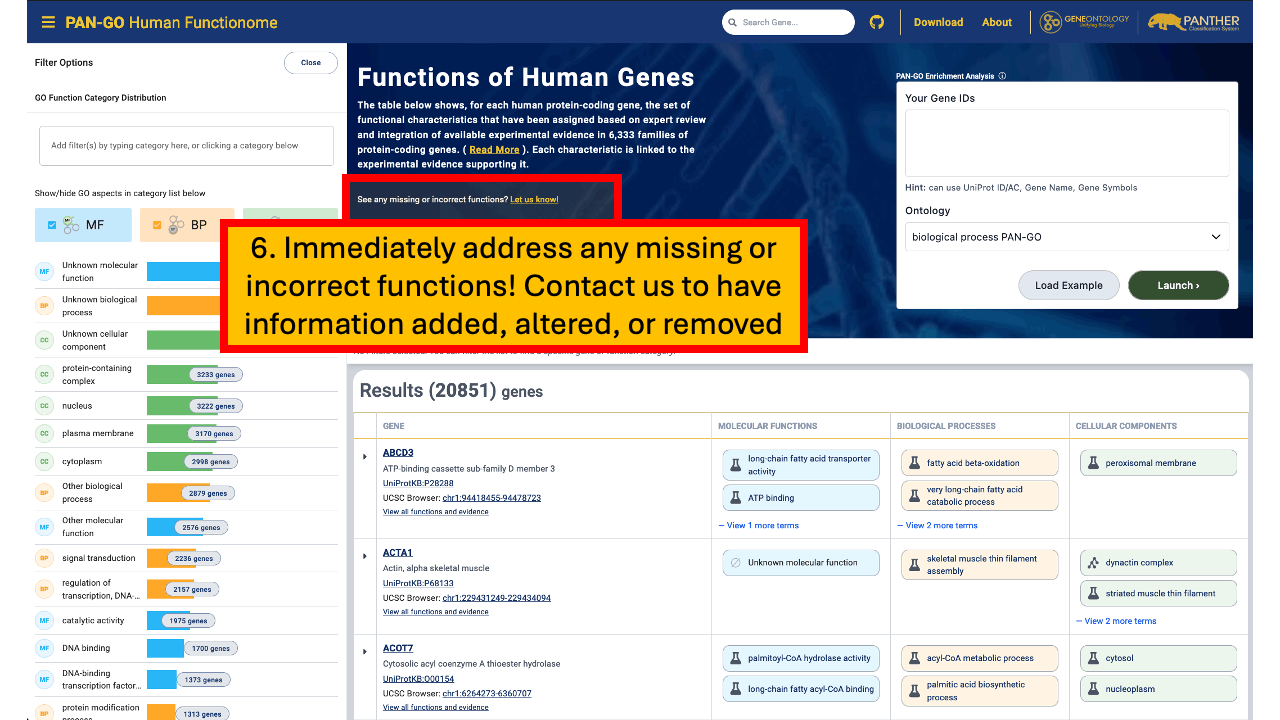
How do I use the GO Function Category Distribution view in the PAN-GO Functionome?
If you want to explore the PAN-GO Functionome by topic (rather than starting from a specific gene), the GO Function Category view lets you browse human genes grouped into broad functional groupings. A function category (also called a GO subset) is a high-level GO concept used to group genes by their annotated functions.
For example, if you filter to the chromosome category, you will see genes with chromosome-related annotations, and you will not see genes that are only annotated to unrelated categories such as cytoskeleton.
To browse by function category, go to the PAN-GO Human Functionome site and use the Distribution of Genes by Function Category interactive graph/filter. You can show or hide GO aspects (MF, BP, CC) and then click one or more categories in the graph to apply filters. The gene table updates to show only genes in the selected category (or categories). You can remove filters to broaden the results again.

How do I find the functions of a specific human gene in the PAN-GO Functionome?
If you already have a gene of interest and want a concise summary of its annotated functions, use the gene search and open the gene’s functionome view.
Go to the PAN-GO Human Functionome site and use the search box to enter a UniProt ID/AC, Gene Name, or Gene Symbol. Select the matching gene from the results.
The results page summarizes the gene’s functional characteristics using GO terms, organized by Molecular Functions, Biological Processes, and Cellular Components. For full details, click the gene name in the table to open the gene’s detailed view, including supporting evidence for each annotation.

AmiGO
How do I browse the GO?
The GO Consortium has developed AmiGO for searching and browsing the Gene Ontology and the gene products that member databases have annotated using GO terms. The quick search field autocompletes gene products and GO Terms. Choosing an auto-completed choice from the drop-down will return the summary page for that gene product or term. Alternatively, terms can be entered by free text and the user will be allowed to choose whether the search will return genes, terms or annotations. For more information on using AmiGO, see the AmiGO help documentation.
Learn more about Retrieving GO Data Using AmiGO, API, Files, and Tools from our chapter in the Gene Ontology Handbook.
Where can I find software to allow me to browse the GO terms and annotations?
You can browse GO terms and annotations using various tools. The GO Consortium supports both AmiGO and QuickGO.
AmiGO was developed for searching and browsing the Gene Ontology and the gene products that member databases have annotated using GO terms. Entering a search term into the quick search menu and choosing an auto-completed choice from the drop-down will return the summary page for that gene product or term. Alternatively terms can be entered by free text and the user will be allowed to choose whether the search will return genes, terms or annotations. For more information on using AmiGO, see the AmiGO help documentation.
Learn more about Retrieving GO Data Using AmiGO, QuickGO, API, Files, and Tools from our chapter in the Gene Ontology Handbook.
What data does AmiGO use? Are there IEAs? If so, which ones?
AmiGO is reloaded approximately once a week. The files currently loaded into the public AmiGO instance can always be seen on the load details page.
AmiGO does currently load full Inferred from Electronic Annotations (IEAs) from UniProt, although this is in development. For a more full discussion of the data loaded into AmiGO, please see the FAQ regarding AmiGO and QuickGO data.
Why does AmiGO display annotations to term X but these annotations aren’t in the GAF file?
Simply put, AmiGO displays annotations made to subclasses by default, while the GAF only contains direct annotations. So an AmiGO search for GO:0004672 protein kinase activity will also list annotations to terms like cAMP-dependent protein kinase regulator activity and even positive regulation of epidermal growth factor-activated receptor activity.
More specifically, AmiGO doesn’t just display subclasses, it uses closure over multiple edge types- part_of, is_a, occurs_in and regulates - to group annotations. This is why you’ll see the Process term positive regulation of epidermal growth factor-activated receptor activity in your results after using AmiGO to look for annotations to the Function term GO:0004672 protein kinase activity.
In order to modify the results in an AmiGO search, use the “GO class (direct)” filter. This will limit the results to only what is annotated directly to your GO term.
How do I find manually annotated gene products only, i.e. how do I sort by evidence code?
Search results can be filtered using the filter menu on the left-hand side of the results page of an AmiGO search. Using the drop-down menu a variety of evidence codes or evidence code combinations can be added or removed to filter the set.
What are the differences between the data available in AmiGO and those on QuickGO?
These are some of the differences between EBI-GOA (QuickGO) and GO Central (AmiGO) when it comes to entities.
GO Central recommends that GAF annotations are made to genes, that is 1:1 equivalents. In GOA (and consequently in QuickGO) annotations are made to proteins, and there may be multiple proteins per gene, sometimes representing different isoforms. You will see this reflected in different numbers for mouse annotations for example.
This is a very important difference, one that users can see when comparing UIs, but more importantly, it is about the underlying datasets and whether a gene-centric or protein-centric worldview is chosen.
Additionally, GO Central omits the majority of the sequences and IEA [electronic] annotations from UniProtKB from the weekly database builds due to the large size of the data set. For those species with a dedicated authoritative database group, such as Drosophila, mouse or Saccharomyces, UniProtKB annotations are collected and submitted by the dedicated group, and hence the UniProtKB IEA annotations for these species do appear in the GO database. As an NHGRI funded knowledgebase, GO Central focuses on annotations that elucidate human genes or genes of relevance to human health in some way. GO Central also includes plants, as well as the 200 genomes of the Quest for Orthologs project. More datasets will be supported depending on available resources.
What is the best way to link into AmiGO?
Please refer to the AmiGO 2 wiki manual.
How do I install AmiGO locally?
Full documentation for downloading and installing AmiGO is available on the GO wiki.
GO-CAMs
What is a GO-CAM?
GO-CAM stands for Gene Ontology Causal Activity Model. GO-CAMs link multiple standard GO annotations into an integrated model of a biological system. More information can be found on the Introduction to GO-CAMs page.
How does the information in GO-CAMs compare to existing pathway databases?
GO-CAMs are causally connected GO annotations. Existing pathway databases are not explicitly geared for performing GO annotation, although some such as Reactome include GO terms.
GO-CAMs differ from databases such as BioModels because the information in a GO-CAM is qualitative, whereas the information in BioModels is quantitative.
GO-CAMs have a different model from many pathway databases, which is shown below:
{kind=link}
The GO-CAM model is activity-centric, in that the molecular activity (i.e. GO molecular function) is the central unit of annotation.
The model allows for standard GO annotations (with no causal connection), or for causally connected annotations. This allows for the capture of partial information in an incremental fashion.
The GO-CAM model is simpler than that used by databases such as Reactome: in a GO-CAM we do not typically capture all the participants in a reaction, together with their stoichiometry. Instead this information is included in the GO term.
We are currently investigating translations between pathway formats such as BioPAX and OpenBEL to GO-CAM. See the Pathways2GO repository for more information.
How do I make my own GO-CAMs in Noctua?
How do I get an account for Noctua?
Noctua is the curation platform developed by the Gene Ontology Consortium to allow curators to create both standard GO annotations and GO Causal Activity Models (GO-CAMs). The Noctua curation platform requires training both for the tool as well as general GO curation. Before we create a Noctua account, we’d like to know what your contributions to GO-CAM would be. Please email us with a brief summary of your research project and how GO-CAM is relevant. Include the biological process(es) and organism(s) you want to make GO-CAM for and the time frame of your project (how long you anticipate your project will run).
Enrichment Analysis
How do I use GO’s Term Enrichment tool?
One of the main uses of the GO is to perform enrichment analysis on gene sets. For example, given a set of genes that are up-regulated under certain conditions, an enrichment analysis will find which GO terms are over-represented (or under-represented) using annotations for that gene set.
Users can perform enrichment analyses directly from the home page of the GOC website. Details about the tool, how to use it, and how to interpret the results are available from the GO Enrichment Analysis page.
How can I do term enrichment analysis for a species that is not present in the list from AmiGO?
PANTHER, which supports the backend of the GO enrichment, provides the list of the species found on the right side of GO website. Besides those 130+ genomes, PANTHER supports another 1100+ genomes from the Reference Proteome project for users to generate GO annotations.
If your organism is not one of the nearly 1000 genomes supported in PANTHER, there are two options:
-
The first option is to contact the Reference Proteome project, and work with them to incorporate the genome in their project. Once that is done, you can use the regular process to generate the GO annotation file.
-
The second option is to score your genomes against the PANTHER HMM library. Read our Nature protocol paper, and find the details in Box 2 of the paper.
Does the Term Enrichment tool have a limit on the number genes in the input file?
Our Term Enrichment tool on the homepage of the Gene Ontology Website cannot handle very large gene lists. The root of the problem is that at a certain number, the input (gene list) is too large for the form; however there is not an exact number at which it fails. One way to solve the problem, should you come across this situation, is to reduce the size of the input file by reducing the number of genes.
Alternatively, annotators can use the PANTHER Term Enrichment tool directly, without AmiGO as an intermediary; this would still be the exact same analysis with the GO data. To perform term enrichment analysis directly from the PANTHER website, visit https://pantherdb.org. Once you upload or paste your gene list, select the ‘Statistical overrepresentation test’ option (in Step3) to perform the term enrichment.
What third-party GO Term Enrichment tool should I use?
I need help with a third-party GO Term Enrichment tool.
We recommend our in-house EA tool, as we can help you with troubleshooting as well as guarantee the data are current. If you are using a third-party tool, ensure they provide you with the version of GO. We cannot provide support for these outside tools, and we have found many tools create inaccurate GO annotations. Therefore, we advise caution with third-party tools. If you do find errors with annotations provided to you by such tools, please provide us with the tool and pertinent details, and we will try to contact the developers.
If you need to use a tool other than our EA/PANTHER, we recommend using the latest version of InterProScan 5. InterProScan is a reliable tool from EMBL-EBI that uses PANTHER as well as HAMAP, NCBIfam, Pfam, PROSITE, and other resources to functionally characterize novel nucleotide or protein sequences from FASTA files. InterProScan can even be used with Galaxy, with some limitations.
Subsets
What are GO subsets? What are GO slims?
GO subsets (also known as GO slims) are cut-down versions of the GO containing a subset of the terms. They are specified by tags within the ontology that indicate if a given term is a member of a particular subset. See more at the guide to GO subsets.
What is “GO slimming” ?
Mapping granular annotations of a set of genes to one or more high-level, broader parent terms is referred to as “GO slimming”. GO slimming is commonly used to report an overview of a genome or to a set of summarize experimental results. GO hosts a number of predefined slim sets including a generic GO slim, and a number of slims/subsets tailored to give good coverage for some well studied/annotated model species. GO slimming will only be successful if the organism of interest has a body of GO annotation in the GO database (either electronic or manual). If your organism of interest has no publicly available GO data refer to the FAQ on annotating a gene set.
How do I create a user defined “GO slim”?
GO hosts a number of predefined Slim sets including a generic GO subset and a number of subsets tailored to give good annotation coverage for several well studied/annotated model species. These existing subsets should be sufficient for most users.
The available GO slimming tools also provide an option to upload your own subset. The generic GO slims or organism specific slims are a good starting point to create your own GO subset. For most applications you usually need to adjust the terms in the slim to represent your results (i.e to reduce the number of terms, or to replace terms in regions of special interest with more specific children). You may find the GO Term Mapper, GO Term Finder, and the GO Term Matrix useful in modifying an existing slim or creating a new slim.
When creating a slim you should still ensure that it covers as many annotated genes in your set as possible. To enable interpretation of your results you should also report how many genes are annotated but not in your subset, and how many genes do not slim (i.e map only to the root node and are therefore ‘unknown’).
Contact the contact the GO for more information.
How do I map a set of annotations to high level GO terms (GO slim)?
You can use the GO Term Mapper provided by the Bioinformatics Group at Princeton’s Lewis-Sigler Institute. This web-based tool takes a list of genes with their detailed GO term annotations and maps them to broader GO slim terms, allowing you to bin your genes into broad functional categories.
The tool allows you to select from the three GO aspects (Molecular Function, Biological Process, or Cellular Component) and choose from available GO slims including the generic slim or organism-specific slims (such as S. cerevisiae or plant slims, which are curated to best represent the biology of those organisms). The tool accepts various gene identifier types and provides output in multiple formats including HTML tables with gene links and frequency counts. Please note that web-based tools may have processing limitations and time constraints.
Read more or download the GO slims here.
When reporting GO slim results, why shouldn’t I display my “GO slim” results as a pie chart?
The numbers provided by a GO slim are an annotation count not a gene product count, and so gene products may be present in more than one bin. Therefore displaying GO slim totals as percentages is not meaningful. For your results to be interpreted fully, you should also report how many genes are annotated but not in your slim, and how many genes do not slim.
Mappings
What are mappings?
The files contain concepts from systems external to GO e.g. Enzyme Commission numbers, SWISS-PROT keywords and TIGR roles, indexed to equivalent GO terms. The mappings are typically made manually; details can be found in the file header. See Mappings to GO for available files.
How do I find the annotations (mappings) for Entrez, NCBI or other IDs?
To search the GO database, a list of Entrez IDs, NCBI IDs, etc. needs to be converted to UniProtKB or model organism database IDs.
UniProt and the Protein Information Resource (PIR) have similar ID mapping tools to help with the conversion:
- http://www.uniprot.org/uploadlists/
- https://proteininformationresource.org/pirwww/search/idmapping.shtml
GO annotations from QuickGO can be filtered for many parameters and provide mappings to several IDs, e.g NCBI or Ensembl gene IDs: http://www.ebi.ac.uk/QuickGO/GAnnotation
What is the UniProt GCRP?
UniProt GCRP stands for Gene-Centric Reference Proteome. It indicates a gene-centered, minimally redundant protein set for an organism, intended to provide one representative UniProtKB protein sequence per protein-coding gene. This helps reduce redundancy that can occur when many very similar genomes or annotations exist (common in bacteria and also seen across other taxa), making downstream analyses more consistent.
GCRP is related to, but distinct from, a Reference proteome: a reference proteome is a representative proteome for a species, while GCRP describes the gene-centric representation strategy (one representative sequence per gene) within a proteome rather than attempting to enumerate all isoforms, variants, or strain-specific sequences.
Why are Interpro2go mappings not updated with GOA releases?
GOA is updated in accordance with the latest data released by its core databases (SWISS-PROT, TrEMBL, InterPro, Ensembl) as well as mappings of SWISS-PROT Keywords, InterPro and Enzyme Commission (EC) terms to GO. Each of GOA’s core databases produces its own releases; for example, InterPro has dependencies on the member databases of InterPro. InterPro2GO is updated at regular intervals but not always in keeping with monthly schedule of GOA releases.
Keep in mind that the Gene Ontology Annotation (GOA) resource (http://www.ebi.ac.uk/GOA) provides evidence-based Gene Ontology (GO) annotations to proteins in the UniProt Knowledgebase (UniProtKB), and is not the same as GOC (the entire GO Consortium, including groups like GOA).
File Formats
What are the file formats used by the Gene Ontology?
Ontology files are available in OBO, OWL, and some files are available in JSON. Refer to the ontology downloads page for information on ontology files.
Annotations are available in GAF (Gene Association File), or by the companion files Gene Product Association Data (GPAD) + Gene Product Information (GPI). For general introduction to the Gene Ontology’s annotation file formats, see GAF 2.2 and GPAD file format/GPI file pages.
What is a GAF file?
A GAF (Gene Association File) is a GO annotation file containing annotations made to the GO by a contributing resource such as FlyBase or Pombase. See more information on the GAF file format guide.
What is a GPAD file?
The GPAD - Gene Product Association File Format - is an alternative means of exchanging annotations from the Gene Association File (GAF). The GPAD format is designed to be more normalized than GAF, and is intended to work in conjunction with a separate format for exchanging gene product information. For details, see the GPAD specification page.
What is a GPI file?
A GPI - Gene Product Information file is used to submit gene and gene product information to the GO Consortium. The GPI specification is here. Please note that annotation information relationships between GO terms and annotations made to them uses GPAD; see details on the GPAD specification page.
What exchange format is used for GO-CAMs?
GO-CAMs include more information than standard GO annotations, so cannot be effectively exchanged using the simple tabular formats used by the GO.
The native representation for GO-CAMs is the Web Ontology Language (OWL). Standard RDF exchange formats such as RDF/XML and Turtle can be used for GO-CAMs.
Individual files can be found on GitHub.
What is an OWL file?
OWL is the acronym for Web Ontology Language, a standard produced by the W3C. GO in OWL is based on a translation from OBO to OWL and is available for download. OWL files can be opened in an editing tool such as Protege.
What is OBO file format?
The OBO file format is one of the formats that the Gene Ontology is made available in. The most recent version is OBO 1.4. The OBO format is designed to be more human readable than XML based formats. GO can be accessed in this format on the Downloads page.
Why are the ontologies initially produced in OBO flat file format instead of XML?
The ontologies are initially produced in the specially designed OBO flat file format. They are converted to XML once a month for the convenience of users who require this facility. Both formats and many others are available in the GO downloads section. We use the OBO flat file format because it is human-readable, and also because the file is much smaller without the XML tags. This means that it is much quicker and easier for the curators to handle the file on a day-to-basis.
How can I generate files in the old GO flat file format?
As of August 1, 2009, the original GO flat file format was deprecated and is no longer be provided by the GO Consortium.
The OBO-Edit project, which used to generate the flat file format, has been mothballed.
Why am I having issues accessing the FTP/CVS/SVN service?
Where is go_daily-termdb.rdf-xml?
I can’t find a file we’ve been downloading for years…
In early 2021, we suspended a few of our long running legacy services. These include:
- FTP
- SVN
- CVS
- ext.geneontology.org
- viewvc.geneontology.org
- archive.geneontology.org (now redirects to release.geneontology.org)
These have been replaced with GitHub, release.geneontology.org, and other services. Some files, including go_daily-termdb.rdf-xml and go_monthly-termdb.rdf-xml, were already several years out-of-date. While the new data pipeline produces the data in those files, it is in a different format.
For current download information, please see:
- http://geneontology.org/docs/download-ontology/
- http://geneontology.org/docs/download-go-annotations/
Please contact us if you cannot locate what you were looking for.
If you are here because your file or source is no longer available, we invite you to follow us on Bluesky (@geneontology.bsky.social), Facebook (/geneontology), GitHub go-announcements repo or join our go-friends email list to stay current with significant changes to the GO knowledgebase.
Database Access
What are the recommended data access policies for your services?
AmiGO and the GO relational database servers are a shared resource and thus we require data mining to be performed in a manner that allows others to utilize this resource at the same time. Any activity that mines the GO database or uses AmiGO must be controlled so that only one request is made at a time. If this is not sufficient, you may download and install the database locally. You can also retrieve all the source files that define the data within the database. More details on the database, including downloads and installation, can be found in the GO database guide.
For more information please contact the GO helpdesk.
What GO annotation file URLs changed in mid 2026? Why?
In June 2026, we updated our file organization and naming system to make GO annotation files more consistent and easier to use. What changed:
| Old URL example | New URL example |
|---|---|
https://current.geneontology.org/annotations/goa_human.gaf.gz |
https://current.geneontology.org/annotations/gaf/HUMAN-uniprot.gaf.gz |
Key changes:
- New URL structure: Files now organized by type (
/gaf,/gpad,/gpifolders) - Standardized naming: Files use UniProt organism codes (e.g.,
HUMAN-uniprot.gaf.gzinstead ofgoa_human.gaf.gz) - Separate species files: Multi-species files from Xenbase & CGD split into individual organism files
- Two options for Model Organisms: MOD files available in both MOD-centric (
DANRE-mod.gaf.gz) and UniProt-centric (DANRE-uniprot.gaf.gz) versions
Migration timeline:
- June 2026: New URLs went live
- July 2026: Old URLs still work (one month grace period)
- July-September 2026: Old URLs redirect to new locations
- After September 2026: Old URLs no longer functional
Need help? Check our URL mapping table or contact us if you need assistance updating your data pipelines.
I want to use the database files but…
The *.txt files must be imported into a MySQL instance using mysqlimport. They are not intended to be loaded into excel or parsed using custom tools. Why are so many of the .txt files empty?
For some exports of the GO database, some tables will necessarily be empty. For example, the termdb dump by design omits any data on genes or gene associations, so the corresponding tables are mepty. MySQL requires a file be present for every table, hence there will be some tables with 0 rows in the .txt files.
What is the best way to obtain the GO annotations for a list of UniProt accession numbers in batch?
With UniProt accession numbers, you can obtain all GO annotations by parsing a GOA gene association file, which are provided in a simple tab-delimited format. These files are available from the GO directory.
The GOA project offers users a number of different files; for example:
- all UniProtKB proteins with GO annotation
- human proteins
- if you were only interested in proteins from a particular species, we also provide non-redundant, species-specific files for human, mouse, rat, zebrafish, chicken, cow and Arabidopsis proteins (these files are created using the International Protein Index (IPI) - which provides a top level guide to the main databases that describe the proteomes of higher eukaryotic organisms)
Further information on the content and format of our gene association files can be found in the ReadMe.
Please contact GOA help for further assistance.
What is the status of the GO MySQL database?
While the GO MySQL database is currently considered to be in “legacy” mode, meaning that there will likely not be any new developments on it, it is a widely used and convenient resource for many types of queries. More information about it can be found in the GO MySQL Database Guide.